Notre charte pour le diagnostic in vitro fait a été mise à jour à la suite de plusieurs modifications de la réglementation DIV. Les Commissions de Biologie Clinique et d’Anatomie Pathologique ainsi que le Collège belge pour l’Hérédité Humaine et les Maladies Rares approuvent cette mise à jour.
« Le lit d'hôpital le moins cher est celui que nous pouvons éviter »
Les maladies cardiovasculaires restent la principale cause de décès en Europe. L'Union européenne élabore actuellement un plan d'action pour changer cette situation. « Si nous voulons vraiment réduire l'impact des maladies cardiovasculaires, nous devons miser beaucoup plus sur la prévention », explique le professeur Damien Gruson. « Et la biologie clinique a un rôle important à jouer à cet égard. »
Nouvelles
Publications
DIV
Le professeur Damien Gruson est chef du service de biochimie médicale des Cliniques Universitaires Saint-Luc et président de la section Technologies émergentes de la Fédération internationale de chimie clinique et de médecine de laboratoire (IFCC). À la lumière des projets européens en matière de stratégie de santé cardiovasculaire et des discussions actives au sein de l'Alliance européenne pour la santé cardiovasculaire (EACH), il partage sa vision du rôle crucial de la biologie clinique dans les soins cardiovasculaires.
Professeur Gruson, pourquoi un plan d'action européen en matière de santé cardiaque est-il nécessaire ?
Plus de 60 millions d'Européens vivent avec une maladie cardiovasculaire, et chaque année, 5 millions de nouveaux cas sont diagnostiqués. Avec 1,7 million de décès, il s'agit de la principale cause de mortalité en Europe. Le coût annuel total s'élève à plus de 280 milliards d'euros.
En Belgique aussi, les chiffres sont alarmants : plus d'un million de patients et plus de 30 000 décès par an. Ce sont des statistiques que nous ne pouvons ignorer. Nous devons de toute urgence nous engager à améliorer les soins cardiaques. Un plan d'action européen peut orienter et soutenir les États membres dans cette voie.
Selon vous, sur quoi ce plan doit-il se concentrer ?
La prévention, la prévention et encore la prévention.
Nous savons depuis longtemps que la prévention et le dépistage précoce font une grande différence. Ce sont les leviers qui permettent d'éviter les hospitalisations et les complications, et ainsi d'épargner des souffrances aux personnes et des coûts à la société. Mais la manière dont nous organisons nos soins et nos budgets est très fortement axée sur les soins curatifs.
Il est plus facile à dire qu'à faire, mais nous n'avons pas d'autre choix. La meilleure façon de maîtriser les dépenses de santé est d'intervenir avant que les soins ne soient nécessaires. Dans le contexte économique actuel, le lit d'hôpital le moins cher reste celui que nous pouvons éviter.
Nous devons donc investir dans la prévention, mais nous préférons ne pas réduire les soins curatifs et nous ne disposons pas de moyens supplémentaires. Comment sortir de cette impasse ?
Il n'y a pas de solution miracle. Il existe des solutions, mais elles demandent du temps et beaucoup de travail. Je reste néanmoins optimiste, les étoiles sont bien alignées.
Nous disposons aujourd’hui d’excellentes technologies qui permettent une évaluation précoce des risques et un diagnostic rapide des maladies cardiovasculaires. Pensez aux technologies omiques – telles que la génomique, la transcriptomique ou la protéomique – et aux techniques de séquençage de nouvelle génération, à l’automatisation complète des laboratoires ou encore aux tests POCT (Point of Care Testing). Ajoutez à cela les avancées dans le domaine de l’intelligence artificielle et du traitement des données, et nous sommes désormais capables de détecter les risques beaucoup plus tôt et avec une précision accrue.
Le défi consiste à intégrer intelligemment ces technologies dans la pratique des soins, dans les parcours de soins et dans leur financement. C'est également un point important au sein du département Technologies émergentes de l'IFCC.
Diagnostic préventif en cardiologie : LP(a) et NT-proBNP
Le professeur Gruson cite deux tests de laboratoire comme bons exemples de prévention en matière de soins cardiaques : la détermination de la LP(a) et des peptides natriurétiques (BNP et NT-proBNP).
La lipoprotéine (a) est un type de mauvais cholestérol génétiquement déterminé qui augmente considérablement le risque de maladies cardiovasculaires. La Société Européenne de Cardiologie a réaffirmé l'importance de ce test dans
ses directives
mises à jour en septembre.
« Il est recommandé de mesurer la LP(a) une fois dans sa vie afin d'affiner son profil de risque cardiovasculaire. Une fois que vous connaissez votre taux, vous pouvez agir de manière proactive, soit en modifiant votre mode de vie, soit en prenant des médicaments. De nouvelles classes thérapeutiques ont déjà prouvé leur efficacité. »
Prévention secondaire
Un deuxième exemple est la détermination des peptides natriurétiques (BNP ou NT-proBNP), qui est utilisée pour le dépistage précoce de l'insuffisance cardiaque. « C'est un bel exemple de prévention secondaire », explique le professeur Gruson. « Chez les personnes atteintes de diabète ou présentant d'autres facteurs de risque, cela nous permet de détecter l'insuffisance cardiaque avant même l'apparition des symptômes. Les médecins peuvent ainsi intervenir à temps et éviter des complications graves ou des hospitalisations. »
Que faut-il pour mettre en œuvre plus largement de telles innovations ?
Relier les données et établir des liens. En soi, les données ne veulent pas dire grand-chose. Ce n'est qu'en combinant les données diagnostiques, cliniques et économiques que nous obtenons une image plus complète de l'impact de certains choix.
Que rapporte, par exemple, un investissement dans un test spécifique en termes de coûts évités pour les hospitalisations, les consultations ou les traitements médicamenteux ? À l'inverse, nous pouvons également comprendre dans quels cas les tests sont peut-être prescrits trop souvent. Le débat ne porte alors plus sur le coût des tests, mais sur l'efficacité des soins.
La théorie semble bonne. Est-il réaliste de la mettre en pratique ?
Certainement. Des pays comme l'Estonie et le Danemark prouvent que c'est possible. Ils ont déjà beaucoup progressé en matière d'interopérabilité des données de santé.
Grâce à des systèmes bien intégrés, les prestataires de soins ont une vue d'ensemble du parcours de soins de leur patient, et pas seulement de leur propre partie. Cela permet de prendre des décisions mieux informées, d'obtenir de meilleurs résultats en matière de soins et de réduire les coûts. Cela rend les soins plus durables, au sens large du terme.
Que voulez-vous dire par « durables » ?
La durabilité comporte plusieurs dimensions.
Il y a la durabilité écologique – au sein de la Fédération européenne de médecine de laboratoire, nous y travaillons via l'initiative « green labs ».
Il y a ensuite la durabilité sociale : comment garantir à tous les patients l'accès à des diagnostics de qualité ? La Belgique a été pionnière en Europe dans ce domaine en remboursant le test NIP à toutes les femmes enceintes. C'est cela, la durabilité sociale dans la pratique.
Enfin, il y a la durabilité économique : comment maintenir nos soins abordables ? Le dépistage précoce permet souvent d'éviter des traitements plus lourds et plus coûteux. Cela rend le système viable à long terme. Les aspects économiques et sociaux sont également étroitement liés : un système financièrement viable rend l'accessibilité sociale possible.
Quel rôle voyez-vous pour la biologie clinique dans ce domaine ?
Un rôle essentiel. Nous rendons visible la biologie invisible et la traduisons en informations utiles pour les médecins. Nous aidons à détecter les risques, à éviter les complications et à mieux adapter les soins.
Nous devons donc vraiment nous défaire de l'idée étroite selon laquelle le diagnostic est un coût. Si un test peut prévenir une complication grave, ce n'est pas un coût, mais un investissement dans la qualité de vie.
Pour ancrer ce rôle de manière structurelle, nous avons besoin de plus d'interopérabilité et d'intégration des données. Nous obtenons ainsi une vision plus holistique, les parcours de soins deviennent plus dynamiques et nous pouvons plus rapidement traduire les nouvelles connaissances scientifiques en directives et en financement. Aujourd'hui, cela prend encore trop souvent du temps. Il subsiste donc un fossé entre ce que nous savons et ce que nous faisons dans la pratique.
Quel serait votre message principal aux décideurs politiques ?
Nous disposons d'outils extrêmement puissants : les tests, la technologie et les preuves scientifiques. Ce dont nous avons besoin, c'est d'une collaboration entre cliniciens, médecins de laboratoire, associations scientifiques et décideurs politiques afin d'apporter une valeur ajoutée au patient. Mais aussi avec les entreprises, car elles connaissent le potentiel technologique et doivent comprendre les besoins cliniques afin de développer des solutions ciblées.
Le patient et nos systèmes de soins en bénéficieront. Cela renforcera également la compétitivité de l'Europe. Si nous ne développons plus rien ici, nous devrons bientôt chercher des solutions ailleurs.
July 1, 2025
Jeroen Poels, AFMPS: « L'IVDR doit être une histoire positive pour tout le monde » (partie 2/2)
La semaine dernière, nous avons discuté avec Jeroen Poels, expert en DIV au sein de l'AFMPS, des exigences plus strictes de l'IVDR et de leur impact administratif sur les entreprises. Aujourd'hui, nous élargissons le débat et examinons la signification de l'IVDR pour les différentes parties prenantes.
Nouvelles
Publications
DIV
Monsieur Poels, la semaine dernière, nous avons conclu sur la charge administrative que représente l'IVDR pour les entreprises. Plus d'administration signifie également des coûts plus élevés pour ces entreprises. L’AFMPS le constate-t-il ?
La transition vers la nouvelle législation ne doit en effet pas être sous-estimée pour les entreprises du secteur des DIV. Pour les entreprises de dispositifs médicaux (MD), la réglementation a également changé avec l'arrivée du règlement sur les dispositifs médicaux (MDR), mais les modifications sont moins radicales que celles prévues par l'IVDR.
« Les entreprises du secteur des DIV sont souvent des PME qui ne disposent pas de moyens considérables. »
De plus, de nombreuses entreprises DIV sont des PME qui ne disposent pas des mêmes moyens que les fabricants MD, plus souvent de grande taille. L'Europe tente d'en tenir compte, par exemple avec le paquet de mesures en faveur des PME (2023) et une stratégie européenne spécifique pour les start-ups et les scale-ups (2025).
En reportant l'entrée en vigueur de l'IVDR et en prévoyant des périodes de transition supplémentaires, l'Europe souhaite donner à toutes les parties prenantes concernées suffisamment de temps pour se mettre en conformité.
La pression sur les entreprises du secteur des DIV reste toutefois réelle. Le renforcement des exigences en matière de preuves cliniques et les contrôles effectués par les organismes notifiés et, éventuellement, par les laboratoires de référence européens entraîneront inévitablement une augmentation des coûts pour les entreprises. Nous entendons donc que les fabricants de DIV évaluent de manière critique leur portefeuille de produits : quels produits souhaitent-ils « transférer » vers le cadre de l'IVDR et lesquels souhaitent-ils abandonner ?
Le renforcement de l'IVDR rend inévitablement l'innovation encore plus coûteuse qu'elle ne l'était déjà. Craignez-vous que l'IVDR freine l'innovation ?
C'est difficile à prédire aujourd'hui, mais j'espère que non. En tout état de cause, l'Europe ne veut pas en arriver là et a créé un groupe de travail chargé d'accélérer les procédures relatives aux « innovations de rupture ».
En tant qu'autorité très appréciée au sein de l'UE, l'AFMPS participe pleinement à cette initiative.
L'AFMPS est en effet une marque internationale forte en Europe. Cela ressort clairement du rôle très actif que vous jouez, entre autres, au sein du Medical Device Coordination Group (MDCG).
Notre administration est en effet très active, et ce dans les 13 groupes de travail du MDCG. Ces groupes de travail traitent tous les aspects pertinents pour la mise en œuvre de l'IVDR et du MDR, tels que la notification et le contrôle des organismes notifiés, la vigilance, le contrôle du marché, les études cliniques et de performance, etc.
« Les autorités compétentes d'autres États membres font régulièrement appel à l'expertise belge en matière de DIV. »
Un groupe de travail se concentre spécifiquement sur l'interprétation et la mise en œuvre homogènes des DIV , et conseille les 12 autres groupes sur des sujets spécifiques aux DIV. Depuis 2022, ce groupe DIV a publié pas moins de 16 documents d'orientation. En collaboration avec l'AFMPS, nous avons contribué à chacun de ces 16 documents, souvent en tant que chef de file. Les autorités compétentes des autres États membres de l'UE reconnaissent cette expertise et sollicitent régulièrement notre avis sur des sujets liés aux DIV.
J'ai d'ailleurs récemment été nommé coprésident du groupe de travail sur les DIV orphelins, qui sont des tests destinés à un très petit groupe de patients. Avec ce groupe de travail, nous examinons comment rendre les DIV orphelins plus facilement et plus rapidement disponibles en Europe.
Nous avons déjà longuement évoqué la pression supplémentaire que l'IVDR impose aux entreprises. Qu'en est-il de la pression sur votre administration ?
La base de données EUDAMED devrait à terme nous aider à alléger une partie de la charge administrative, mais ce n'est pas encore le cas aujourd'hui. Entre-temps, de nombreuses tâches ont été élargies et nous en avons reçu de nouvelles. Donc oui, l'IVDR entraîne également une pression supplémentaire pour nous, surtout à court terme.
Dans le même temps, il y a des signes positifs importants. On a longtemps craint, à juste titre, que la capacité des organismes notifiés ne soit pas suffisante pour l'IVDR, le nombre de DIV devant faire l'objet d'une évaluation de conformité étant passé de 20 à 80 % (voir également la première partie de l'interview). Mais ce problème semble se résoudre.
Sur les 17 organismes notifiés pour l'IVDR, seuls deux ne prennent plus de nouveaux clients, selon une enquête récente. L'avant-dernier organisme notifié à figurer sur la liste est SGS Belgium. Notre pays dispose donc désormais d'un organisme notifié pour l'IVDR, ce qui est évidemment une bonne nouvelle pour les fabricants belges !
Nous constatons également une augmentation du nombre de demandes d'études de performance. Il s'agit souvent d'études combinées avec un médicament. Le test DIV sert alors à sélectionner les patients pour l'essai et peut être commercialisé ultérieurement comme diagnostic compagnon. Un grand projet européen, COMBINE, est actuellement en cours afin de simplifier et d'harmoniser les demandes d'études combinées.
Ces signaux sont très importants. Après tout, l'IVDR doit être une réussite.
Absolument, en premier lieu pour les patients et la santé publique.
Les avantages potentiels de l'IVDR pour les patients sont évidents :Plus les tests de diagnostic in vitro sont fiables et efficaces, plus ils bénéficient au patient . Et grâce à une meilleure traçabilité, les dispositifs posant problème peuvent être localisés de manière très précise.
« Plus les tests de diagnostic in vitro sont fiables et efficaces, plus ils bénéficient au patient»
Toutes les mesures prévues par l'IVDR y contribuent plus ou moins grandement. Il est inévitable qu'un changement réglementaire d'une telle ampleur soulève de nombreuses questions. Mais celles-ci ne doivent pas remettre en cause l'ensemble de la réforme.
Il est important d'évaluer l'IVDR de manière continue et rigoureuse et d'oser l'ajuster si nécessaire : que peut-on simplifier, quelles exigences offrent trop peu de valeur ajoutée et peuvent donc être modifiées ou supprimées, etc. L'Europe recherche activement ce retour d'information. Ainsi, une consultation publique des parties prenantes sur l'IVDR et le MDR s'est achevée fin mars et ses résultats sont actuellement en cours d'analyse. Nous continuons également à recueillir les commentaires des parties prenantes concernées en Belgique par l'intermédiaire de l'AFMPS.
June 24, 2025
Jeroen Poels, AFMPS: « Les entreprises peuvent être nos antennes » (partie 1/2)
Depuis la fin du mois dernier, l'IVDR est en vigueur depuis 3 ans. Comment l'AFMPS a-t-elle vécu cette nouvelle réglementation jusqu'à présent ? Qu'est-ce qui fonctionne bien et qu'est-ce qui peut être amélioré ? Nous avons discuté avec Jeroen Poels, expert en DIV au sein de l'AFMPS. « Plus nous détectons tôt les problèmes potentiels liés à l'IVDR, plus nous pouvons agir. Mais pour cela, nous avons besoin de l'aide des entreprises. »
Nouvelles
Publications
DIV
Le règlement européen sur les dispositifs médicaux de diagnostic in vitro, ou IVDR, est applicable depuis le 26 mai 2022. Il remplace la directive sur les dispositifs médicaux de diagnostic in vitro (IVDD). Son objectif ? Améliorer la sécurité et la qualité des dispositifs médicaux de diagnostic in vitro (DIV), renforcer la transparence et la traçabilité des DIV et harmoniser la mise en œuvre de la législation au sein de l'Union Européenne (UE)
Dans une interview en deux parties, Jeroen Poels décortique l'IVDR. Il est expert en dispositifs médicaux pour le diagnostic in vitro à l'Agence fédérale des médicaments et des produits de santé (AFMPS), qui veille à la sécurité, à la qualité et à l'efficacité des dispositifs médicaux sur le marché belge.
Monsieur Poels, commençons par le commencement. Pourquoi fallait-il remplacer la directive IVDD ?
L'IVDD datait de 1998, à une époque où les possibilités diagnostiques étaient beaucoup plus limitées qu'aujourd'hui. À l'époque, par exemple, les tests génétiques, les tests au chevet du patient (point of care tests ou POCT), les logiciels DIV, etc. n'existaient pratiquement pas. Afin de cadrer correctement toutes ces nouvelles possibilités diagnostiques, souvent complexes, et d'anticiper l’apparition de nouveaux tests diagnostiques, la réglementation devait être entièrement repensée.
Lors de l'élaboration d'un nouveau cadre, l'Europe a délibérément opté pour un règlement plutôt qu'une directive. Une directive peut être transposée dans la législation nationale de chaque État membre. Cela offre une certaine flexibilité. Cependant, les défis liés à la sécurité et à la qualité des dispositifs médicaux de diagnostic dépassent les frontières nationales, d’où le choix d'un règlement, qui doit être appliqué de manière quasi identique par tous les États membres.
Quelles sont les principales nouveautés par rapport à la directive IVDD ?
L'IVDR compte 160 pages, contre 37 pour l'IVDD. Les différences sont donc nombreuses. Les plus importantes concernent les exigences auxquelles doivent satisfaire les DIV et leur contrôle.
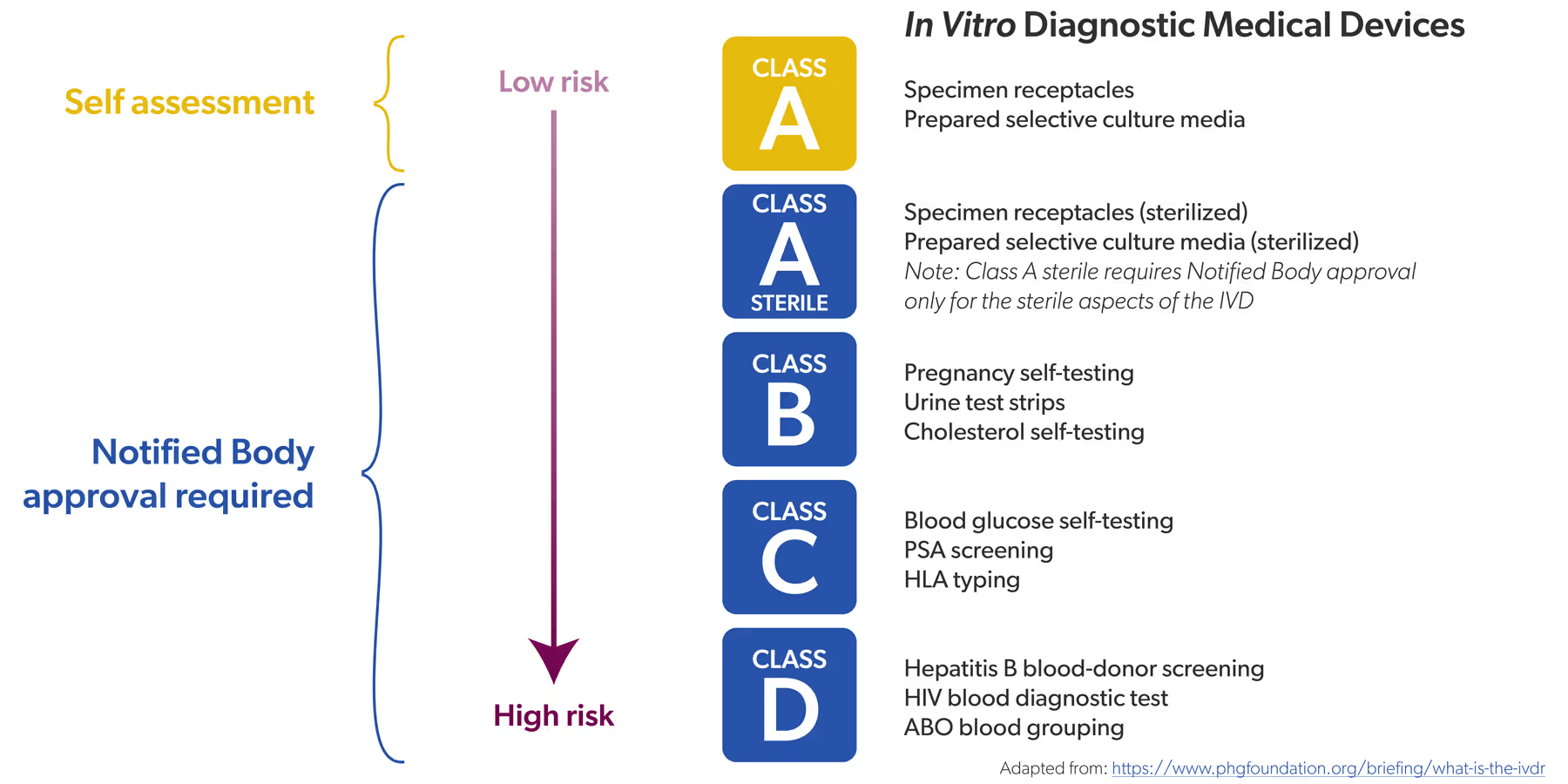
Auparavant, sous l'IVDD, un nombre limité de DIV étaient classés comme « à haut risque ». Il s'agissait par exemple des tests de dépistage du VIH ou de l'hépatite. Ces DIV devaient faire l'objet d'une évaluation de conformité par un organisme notifié – cela concernait environ 20 % de tous les DIV – avant de pouvoir être mis sur le marché. Pour les 80 % restants, une auto certification par le fabricant suffisait.
« Auparavant, 20 % de tous les DIV devaient être contrôlés par un organisme notifié, contre 80 % aujourd'hui. »
Dans le cadre de l’IVDR, tous les DIV ont reçu une classe de risque (de faible à élevée, de A à D) et le rapport s'est inversé : environ 80 % de tous les DIV doivent désormais être contrôlés par un organisme notifié, contre 20 % auparavant. Seuls les DIV non stériles de classe A peuvent encore faire l'objet d'une autocertification, par exemple les réactifs de couleur.
Les évaluations de conformité sont également plus strictes, tout comme la surveillance des organismes notifiés qui les effectuent. Pour les DIV à haut risque, il existe désormais des acteurs supplémentaires qui participent à l'évaluation de la conformité, tels que le comité d'experts en DIV et les laboratoires de référence européens. Leur expertise est un atout important.
Concrètement, qu'implique une évaluation de conformité plus stricte ?
Les exigences en matière de sécurité des produits, d'identification et de traçabilité ont été considérablement renforcées. La barre est également placée beaucoup plus haut en termes de performances. Avant qu'un DIV puisse être mis sur le marché, il faut désormais fournir beaucoup plus de preuves cliniques de l'efficacité du dispositif.
Sous l'IVDD, une déclaration du fabricant indiquant qu'une étude de performance serait réalisée suffisait. L'IVDR impose une charge de la preuve beaucoup plus importante, en particulier pour les études présentant un risque plus élevé pour les patients participants.
Toute personne souhaitant mener une telle étude doit en outre obtenir l’accord préalable d’un comité d’éthique et de l’autorité compétente.
« Un projet pilote européen visant à simplifier la procédure de demande pour les études multinationales a récemment été lancé. »
Parallèlement, l'Europe cherche des moyens de réduire la charge administrative. Dans ce contexte, un projet pilote a récemment été lancé, dans le cadre duquel les promoteurs qui souhaitent mener une étude dans plusieurs États membres pourront introduire une demande unique. L'évaluation de cette demande sera coordonnée sous la direction d'une seule autorité compétente, plutôt que par chaque État membre séparément.
En matière de charge administrative, MedTech Europe met en garde contre les difficultés que ces exigences supplémentaires font peser sur les entreprises, qui risquent de rendre certains DIVindisponibles dans l'UE. En Belgique, nous ne constatons pour l'instant aucun problème, mais nous surveillons la situation rigoureusement. Quel est le point de vue de l'AFMPS à ce sujet ?
En tant que pouvoirs publics, nous sommes bien conscients des risques potentiels et suivons la situation de près. L’UE en est également consciente. C'est pourquoi elle a par exemple prolongé les périodes de transition pour les DIV déjà commercialisés sous la directive IVDD. Afin d'avoir une meilleure vue d'ensemble des pénuries éventuelles, elle a récemment introduit l'obligation pour les fabricants de signaler à l'avance aux autorités et aux clients les pénuries prévues de dispositifs médicaux importants.
En Belgique, nous ne constatons effectivement aucun problème urgent. À l'heure actuelle, nous n'avons connaissance que d'un test de dépistage de la syphilis important, fabriqué par une entreprise asiatique, qui ne sera plus commercialisé en Europe. Mais la Belgique est avant tout un pays de distribution : les fabricants de DIV sont principalement actifs dans d'autres pays. Par conséquent, nous ne sommes généralement pas les premiers informés lorsqu'un fabricant décide d'arrêter la production d'un test.
Notre appel à beMedTech et à vos membres est donc clair : si vous sentez que quelque chose se prépare, avant que cela ne devienne un véritable problème, n'hésitez pas à nous en faire part. Nous mettrons alors tout en œuvre pour trouver des solutions ensemble.
C'est comme pour les problèmes de santé : plus on intervient tôt, plus on a de chances d'éviter une aggravation. Vos membres sont les oreilles dont nous avons besoin pour détecter les problèmes potentiels le plus tôt possible.
Paramètres des cookies
Nous utilisons des cookies pour assurer le bon fonctionnement de notre site web et pour mesurer l’utilisation ainsi que l’efficacité de notre marketing. En acceptant, vous acceptez notre politique de confidentialité.
Nous utilisons des cookies pour assurer le bon fonctionnement de notre site web et pour mesurer l’utilisation ainsi que l’efficacité de notre marketing. En acceptant, vous acceptez notre politique de confidentialité.